

Our
Research

Research at the Rabadan Lab
Our main scientific interests lie in modeling and understanding the dynamics of biological systems through the lens of genomics. We are a very interdisciplinary team of mathematicians, physicists, engineers, biologists, and medical doctors with a common goal of solving pressing medical problems. We are currently focusing our work on:
Cancer. Genomic technologies provide an extraordinary opportunity to identify mutations that contribute to the development of tumors. We are mapping the evolution of cancers and uncovering the mechanisms of response or lack of response to multiple therapies. We work with clinicians and experimentalists all around the world.
Infectious diseases. Evolution is a dynamic process that shapes genomes. Our team at Columbia is developing algorithms to analyze genomic data, with a view to understanding the molecular biology, population genetics, phylogeny, and epidemiology of viruses. We are interested in the emergence of infectious diseases, pandemics and uncovering the mechanisms of adaptation of viruses to humans.
Electronic Health Records. Clinical databases constitute a rich and complex source of raw data. We are using the power of statistics and computers to tease out important clinical patterns in these diverse, important datasets. Combining molecular and clinical data illuminates some of the mechanisms underlying complex diseases.
In particular, we develop mathematical, statistical, and computational approaches, which cover the analysis of high throughput data right through to the altogether more abstract identification of global patterns in evolutionary processes. Learn more about the Rabadan Lab and the three main global questions that we are addressing.
Research Projects
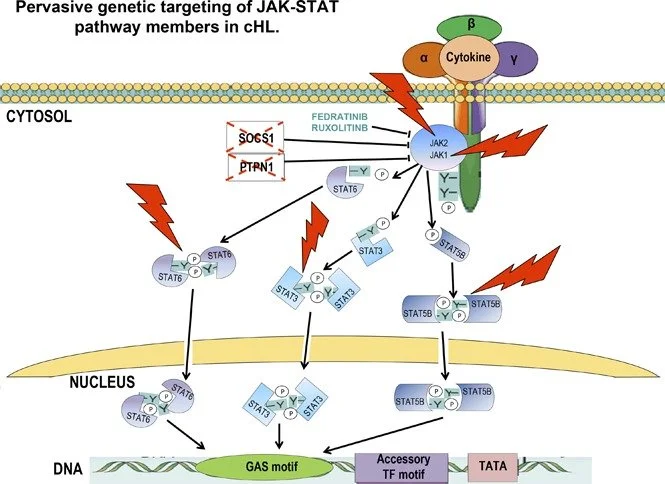
Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma
Dissecting the pathogenesis of classical Hodgkin lymphoma (cHL), a common cancer in young adults, remains challenging because of the rarity of tumor cells in involved tissues (usually lower than 5%). Here, we analyzed the coding genome of cHL by microdissecting tumor and normal cells from 34 patient biopsies for a total of ∼50 000 singly isolated lymphoma cells. We uncovered several recurrently mutated genes, namely, STAT6 (32% of cases), GNA13 (24%), XPO1 (18%), and ITPKB (16%), and document the functional role of mutant STAT6 in sustaining tumor cell viability. Mutations of STAT6 genetically and functionally cooperated with disruption of SOCS1, a JAK-STAT pathway inhibitor, to promote cHL growth. Overall, 87% of cases showed dysregulation of the JAK-STAT pathway by genetic alterations in multiple genes (also including STAT3, STAT5B, JAK1, JAK2, and PTPN1), attesting to the pivotal role of this pathway in cHL pathogenesis and highlighting its potential as a new therapeutic target in this disease.

Comprehensive characterisation of compartment-specific long non-coding RNAs associated with pancreatic ductal adenocarcinoma
We developed a computational framework to reconstruct the non-coding transcriptome from crosssectional RNA-Seq, integrating somatic copy number alterations (SCNA), common germline variants associated to PDA risk and clinical outcome. We generated a catalogue of PDA-associated lncRNAs. We showed that lncRNAs define molecular subtypes with biological and clinical significance. We identified lncRNAs in genomic regions with SCNA and single nucleotide polymorphisms associated with lifetime risk of PDA and associated with clinical outcome using genomic and clinical data in PDA. We found that loss of LINC00673 regulates the epithelial differentiation state in PDA cells, increases migratory capacity in vitro and in vivo, and results in loss of epithelial and gain of mesenchymal markers, both in vitro and in tumour samples. This finding is further reflected in poor clinical outcome in low LINC00673 tumours. We expect that the collection of PDA-associated lncRNAs will aid in the design of targeted therapies and may contribute to the development of improved diagnostic tools for PDA. The recent clinical approval of the first antisense therapy for human disease provides a viable, practical approach for leveraging this new understanding of cancer biology.

Geometry and topology of genomic data
The Handbook of Discrete and Computational Geometry is intended as a reference book fully accessible to nonspecialists as well as specialists, covering all major aspects of both fields. The book offers the most important results and methods in discrete and computational geometry to those who use them in their work, both in the academic world - as researchers in mathematics and computer science - and in the professional world - as practitioners in - fields as diverse as operations research, molecular biology, and robotics. Discrete geometry has contributed significantly to the growth of discrete mathematics in recent years. This has been fueled partly by the advent of powerful computers and by the recent explosion of activity in the relatively young field of computational geometry. This synthesis between discrete and computational geometry lies at the heart of this Handbook. A growing list of application fields includes combinatorial optimization, computer-aided design, computer graphics, crystallography, data analysis, error-correcting codes, geographic information systems, motion planning, operations research, pattern recognition, robotics, solid modeling, and tomography.
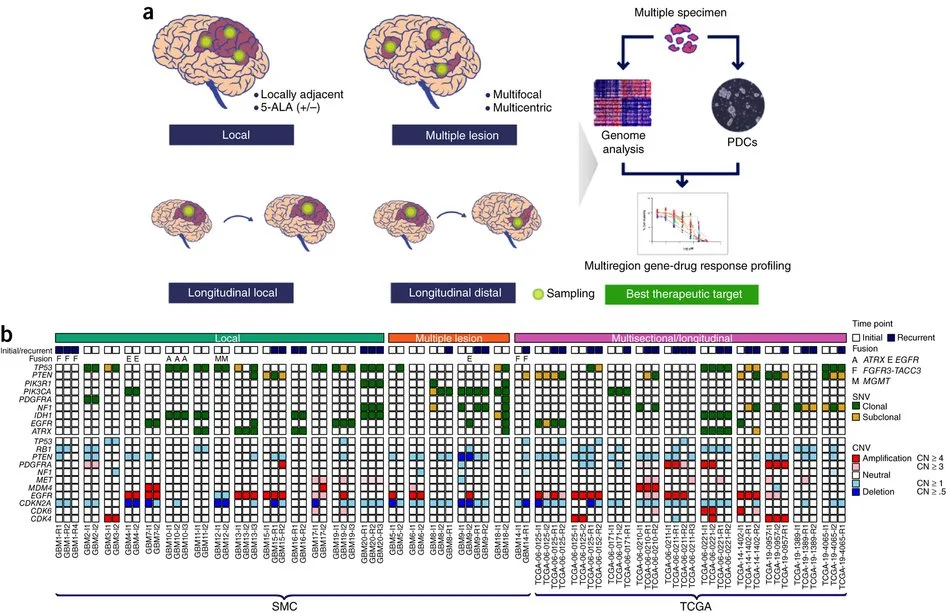
Spatiotemporal genomic architecture informs precision oncology in glioblastoma
Precision medicine in cancer proposes that genomic characterization of tumors can inform personalized targeted therapies. However, this proposition is complicated by spatial and temporal heterogeneity. Here we study genomic and expression profiles across 127 multisector or longitudinal specimens from 52 individuals with glioblastoma (GBM). Using bulk and single-cell data, we find that samples from the same tumor mass share genomic and expression signatures, whereas geographically separated, multifocal tumors and/or long-term recurrent tumors are seeded from different clones. Chemical screening of patient-derived glioma cells (PDCs) shows that therapeutic response is associated with genetic similarity, and multifocal tumors that are enriched with PIK3CA mutations have a heterogeneous drug-response pattern. We show that targeting truncal events is more efficacious than targeting private events in reducing the tumor burden. In summary, this work demonstrates that evolutionary inference from integrated genomic analysis in multisector biopsies can inform targeted therapeutic interventions for patients with GBM.

Evolutionary history of deadly brain tumor
Glioblastoma (GBM) is the most common and most aggressive brain tumor in adults. Current treatment involves surgery, radiotherapy, and chemotherapy plus alkylation agents. Although intensively treated, GBM will always recur. The recurrent tumor will be typically resistant to therapy, leading to death. To understand how GBM evolves under therapy, we have analyzed longitudinal genomic/transcriptomic data from 114 patients, and uncovered the evolutionary landscape of GBM. Importantly, we found 63% of patients experience expression-based subtype changes, 15% of tumors present hypermutation at relapse in highly expressed genes, and 11% of recurrence tumors harbor mutations in LTBP4, which encodes a protein binding to TGF-β.

Topological data analysis captures recombination from large genomic samples
Population-based recombination maps capture the recombination history of populations using genomic data and are a valuable tool in the study of human recombination. We have developed fast statistical estimators of the recombination rate based on topological summaries. Compared to standard linkage-based estimators, topology-based estimators can deal with a larger number of segregating sites and genomes without incurring excessive computational costs. Applying these estimators to phased genotype data of 647 human individuals, we have produced high-resolution, genome-wide maps of human recombination, which have uncovered several novel associations. Specific transcription factor binding sites are frequently associated with recombination. These include binding sites of MLL complexes, which play prominent regulatory roles in germ cell development and early embryogenesis. Additionally, some repeat-derived loci, coding families of transposable elements that are expressed during embryogenesis, are also enriched for recombination.

Reprogramming eukaryotic translation with ligand-responsive synthetic RNA switches
Protein synthesis in eukaryotes is regulated by diverse reprogramming mechanisms that expand the coding capacity of individual genes. One such mechanism is programmed ribosomal frameshifting (PRF). In this work, efficient PRF stimulatory RNA elements were discovered by in vitro selection, and then ligand-responsive switches were constructed by coupling PRF stimulatory elements to RNA aptamers using rational design and directed evolution. Motif discovery was enabled by the methodological novelty of deep sequencing an initially randomized library of RNA sharing a certain pseudoknot scaffold that had undergone multiple rounds of in vitro selection for PRF. This approach led to a rich characterization of precise pseudoknot geometries that can facilitate translation reprogramming, an area with great potential for synthetic biology.
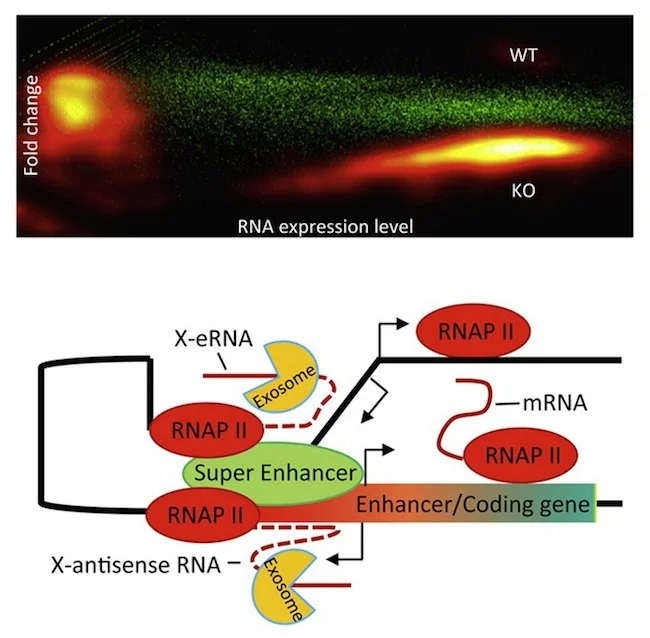
Identifying Novel Noncoding RNAs
The human genome project has shown that only a small fraction (<2%) of human genome can be transcribed into mRNA that is further translated into protein, and the vast majority of the mammalian genome might express non-coding RNA (ncRNA). Although a number of long non-coding RNAs (lncRNAs) have been recently shown to play significant roles in the regulation of gene expression or protein activity in critical signaling pathways, the total number of ncRNAs and the fraction of functional ncRNAs within the mammalian genome are still mysteries. To reveal the landscape of ncRNA expression and specifically, to capture the expression of transient RNAs, we have developed an RNA-seq Analysis pipeline of Transcriptome Reconstruction and Annotation to Identify Novel non-coding RNAs from exosome deficient cells (ATRAIN).

Connections between Mendelian Diseases and Cancer
"If germline genetic variation in Mendelian loci predisposes bearers to common cancers, the same loci may harbour cancer-associated somatic variation. Compilations of clinical records spanning over 100 million patients provide an unprecedented opportunity to assess clinical associations between Mendelian diseases and cancers. We systematically compare these comorbidities against recurrent somatic mutations from more than 5,000 patients across many cancers. Using multiple measures of genetic similarity, we show that a Mendelian disease and comorbid cancer indeed have genetic alterations of significant functional similarity."
---Nature Communications

Non-Hodgkin’s Lymphoma
"The first-ever systematic study of the genomes of patients with ALK-negative anaplastic large cell lymphoma (ALCL), a particularly aggressive form of non-Hodgkin’s lymphoma (NHL), shows that many cases of the disease are driven by alterations in the JAK/STAT3 cell signaling pathway. The study also demonstrates, in mice implanted with human-derived ALCL tumors, that the disease can be inhibited by compounds that target this pathway, raising hopes that more effective treatments might soon be developed."
---CUMC Newsroom
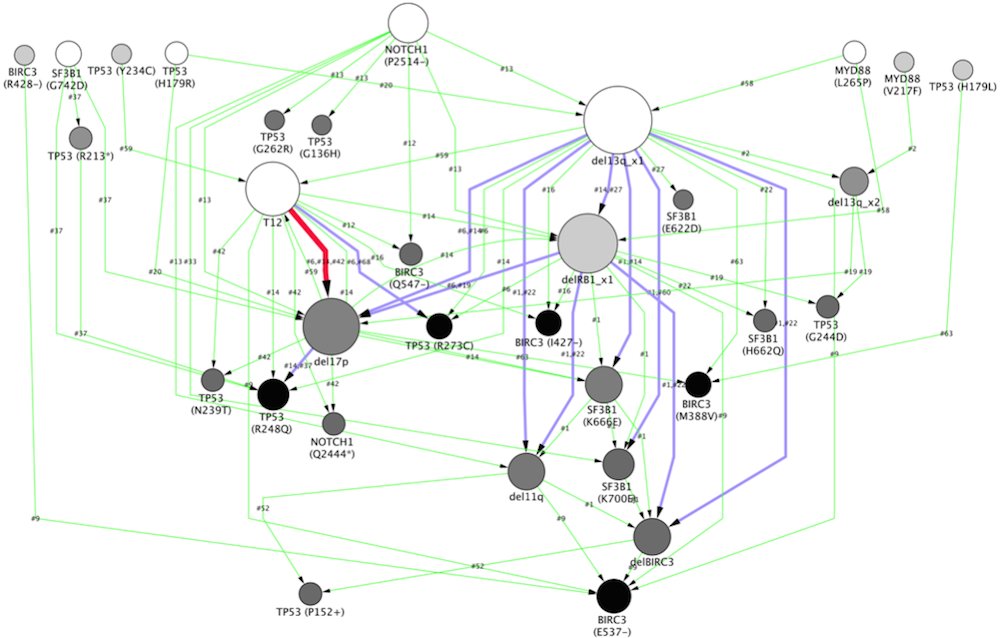
Chronic Lymphocytic Leukemia
A graph representing the sequence of genomic alterations in chronic lymphocytic leukemia (CLL). Each node represents a mutation, with arrows indicating temporal relationships between them. The size of the nodes indicates the number of patients in the study who exhibited the alteration, while the thickness of the lines shows how often the temporal relationships between nodes were seen. The method the researchers use enabled them to identify multiple, distinct evolutionary patterns in CLL.

Identifying Novel Noncoding RNAs
Activation-induced cytidine deaminase (AID) is an enzyme that generates mutations and translocations in mature B cells to produce antibody diversity by targeting immunoglobulin loci, but “off-targets” of AID also lead to cancer. The mechanism of how AID finds its targets is still unclear. By conditionally knocking out a protein Exosc3 in the RNA exosome complex, we have identified a novel type of noncoding RNA, xTSS-RNA, which is most strongly expressed at genes that accumulate AID-mediated somatic mutations and/or are frequent translocation partners of DNA double-stranded breaks generated at the immunoglobulin heavy chain (IgH), indicating a role of this noncoding RNA in the AID targeting mechanism.
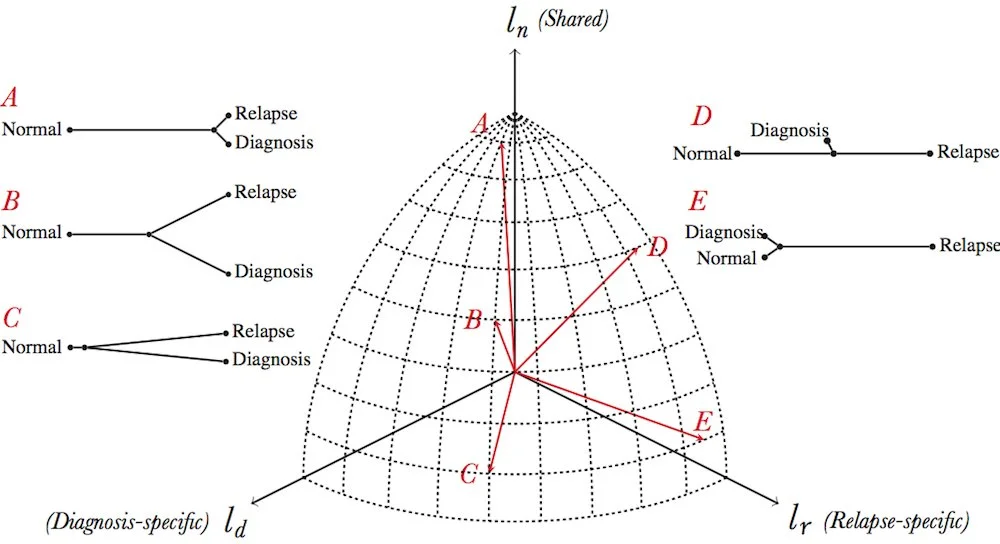
Tumor Evolution
Tumor evolutionary modes visualized in PΣ3.
A: frozen evolution
B: branched evolution
C: divergent evolution
D: linear evolution
E: somatic hypermutation
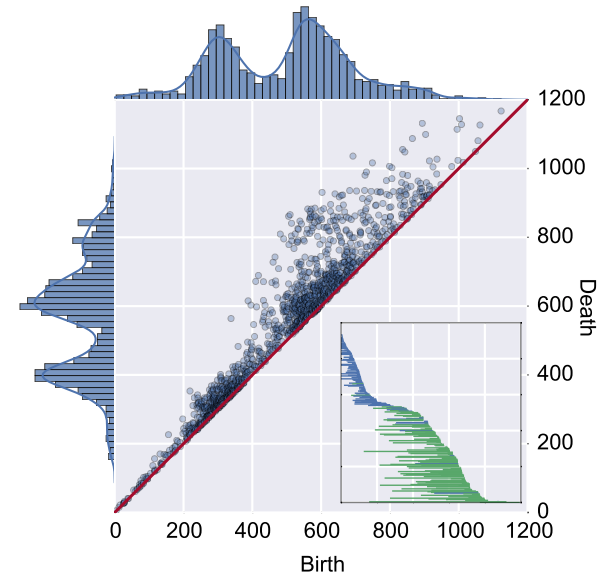
Parametric Inference using Persistence Diagrams: A Case Study in Population Genetics
Persistent homology computes topological invariants from point cloud data. Recent work has focused on developing statistical methods for data analysis in this framework. We show that, in certain models, parametric inference can be performed using statistics defined on the computed invariants. We develop this idea with a model from population genetics, the coalescent with recombination. We apply our model to an influenza dataset, identifying two scales of topological structure which have a distinct biological interpretation.

Bacterial Evolution
Topological network representation of S. aureus genome profiles. Color corresponds to enrichment in mecA, an antibiotic resistance gene.

A Topological Approach to Modeling Evolution
"Recent genomic studies have made it clear that evolution does not only proceed in a 'vertical' pattern in which one organism inherits genomic information from the organisms from which it descends (figure A). Scientists now understand that genomic evolution can also be 'horizontal'; that is, genomic information can be transferred between organisms or evolutionarily similar groups of organisms that are in parallel lineages (figure B), such as in cases of species hybridization in eukaryotes, lateral gene transfer in bacteria, recombination and reassortment in viruses, viral integration in eukaryotes, and fusion of genomes of symbiotic species. These observations suggest that phylogenetic trees have limitations in their ability to characterize evolution at the molecular level and that another model is needed that can integrate both vertical and horizontal evolution.”
---CU Systems Biology News